|
Th1 lymphocytes isolated from the lungs of patients with
severe COVID-19 symptoms have an intracrine (https://vitamindstopscovid.info/02-intracrine/) signaling pathway
which should be
activated by high levels of complement (WP), to turn these cells off their initial hyper-inflammatory
program which produces pro-inflammatory IFNγ (interferon_gamma WP
which has antiviral and anti-bacterial activity as well as stimulating
inflammation: cell destruction such as by natural killer cells WP) and instead cause them to produce the anti-inflammatory cytokine IL-10.
(The cells always produce both these cytokines, but this transition to
a shutdown, anti-inflammatory program, involves less IFNγ and a lot
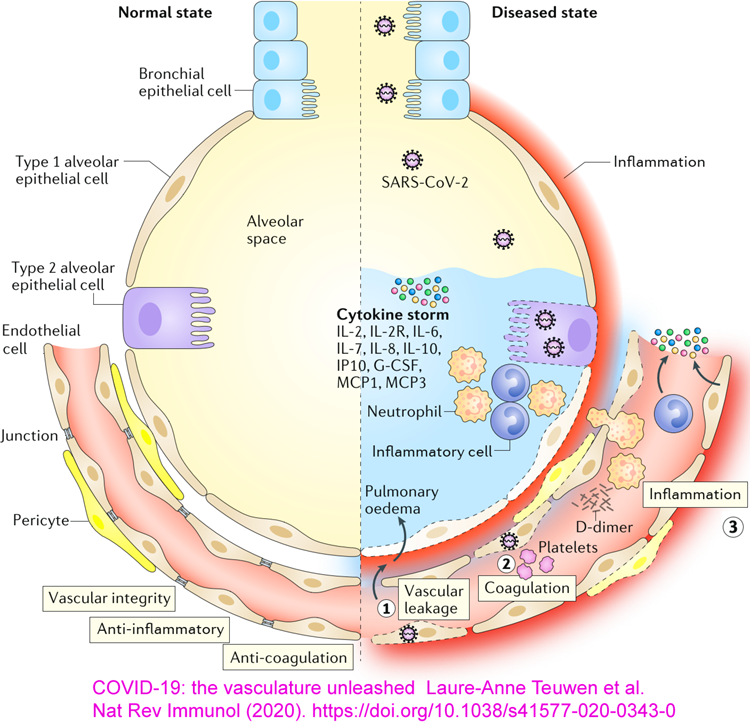
more IL-10.) This is a molecular and cellular explanation for why people with low 25-hydroxyvitamin D have self-destructive, wildly dysregulated, indiscriminate cell destroying, overly-inflammatory immune responses. Such responses drive sepsis, severe influenza, Kawasaki disease (KD WP), Multisystem Inflammatory Syndrome (MIS discussion) and of course severe COVID-19. (See Paul Marik's explanation https://www.evms.edu/covid-19/covid_care_for_clinicians/ of how it is the immune response, not the virus, which causes the escalation to severe symptoms and death. See ../#2015-Stagi for research which shows KD children have very low 25(OH)D vitamin D levels.) In severe COVID-19, severe inflammation in the lungs damages
endothelial cells (the inner lining of blood vessels and capillaries WP)
leading to hypercoagulative blood, causing microembolisms and larger
clots all over the body, which cause most of hypoxia, lasting harm and
death. It is not known whether the cause of all the hyper-inflammatory immune system dysregulation - which causes some COVID-19 sufferers people to develop severe symptoms - is primarily the failure of these Th1 lymphocytes to switch from being pro- to anti-inflammatory, or whether this endothelial cell destruction etc. is also driven to a significant degree by similar failures in the intracrine signaling systems of many other types of regulatory and/or directly anti-pathogen immune cell and/or by direct actions of the virus. However, the determination of the exact mechanism of failure in Th1 cells, in the context of such failures likely also occurring in other cell types, is an extraordinarily valuable contribution which deserves to be very widely known. Low
circulating 25-hydroxyvitamin D (produced in the liver
from UV-B-generated and/or ingested vitamin D3 cholecalciferol) levels are
well known to reduce the effectiveness of numerous direct,
anti-pathogen, responses by the innate immune system cells and to
hinder the creation of antibodies for adaptive immune responses.
These
immune functions of 25-hydroxyvitamin D are due to it being needed, in the
circulation, at higher levels than are sufficient for bone health
(sufficient for the kidneys to produce their much lower concentration
of circulating - and so hormonal - 1,25-dihyroxyvitamin D), to
supply the intracrine / paracrine (inside the cell / to nearby cells)
signaling systems of many or perhaps all types of immune cells. All types of
immune
cell can express the vitamin D receptor - and this is for
intracrine/paracrine signaling - not for responding to the much lower
levels of circulating 1,25-dihydroxyvitamin D which regulates
calcium-bone metabolism. https://vitamindstopscovid.info/02-intracrine/#02-nothorm . See http://aminotheory.com/cv19/#2020-Fabbri [B] for why 40ng/ml or more 25OHD is required for these autocrine signaling systems to function properly. See also the Quraishi et al. graph https://vitamindstopscovid.info/02-intracrine/#04-quraishi which suggests that innate immune cell responses which fight bacterial and perhaps fungal infections keep improving, presumably due to faster and stronger intracrine/paracrine signaling, as 25-hydroxyvitaminD levels rise, up to about 55ng/ml. Please also see ../#25plusD3 for my suggestion of oral calcifediol (25-hydroxyvitamin D) plus vitamin D3 as the best treatment for hospitalised COVID-19 patients, since this raises circulating 25-hydroxyvitamin D to the levels needed for intracrine / paracrine signaling in a few hours, rather than in the several days to a week with, for instance, a 10 mg 400,000 IU bolus dose of vitamin D3. See this 0.014 mg calcifediol per kg body weight recommendation in Prof. Wimalawansa's article in Nature, in 2022: https://www.mdpi.com/2072-6643/14/14/2997.Very strong clinical evidence of the importance of rapidly raising circulating 25OHD levels hospitalised COVID-19 patients can be found the Cordoba calcifediol (25OHD) RCT: Castillo et al. 2020: ../#2020-Castillo . |
!! The graphic below refers to "autocrine" but it should be "intracrine" signaling.
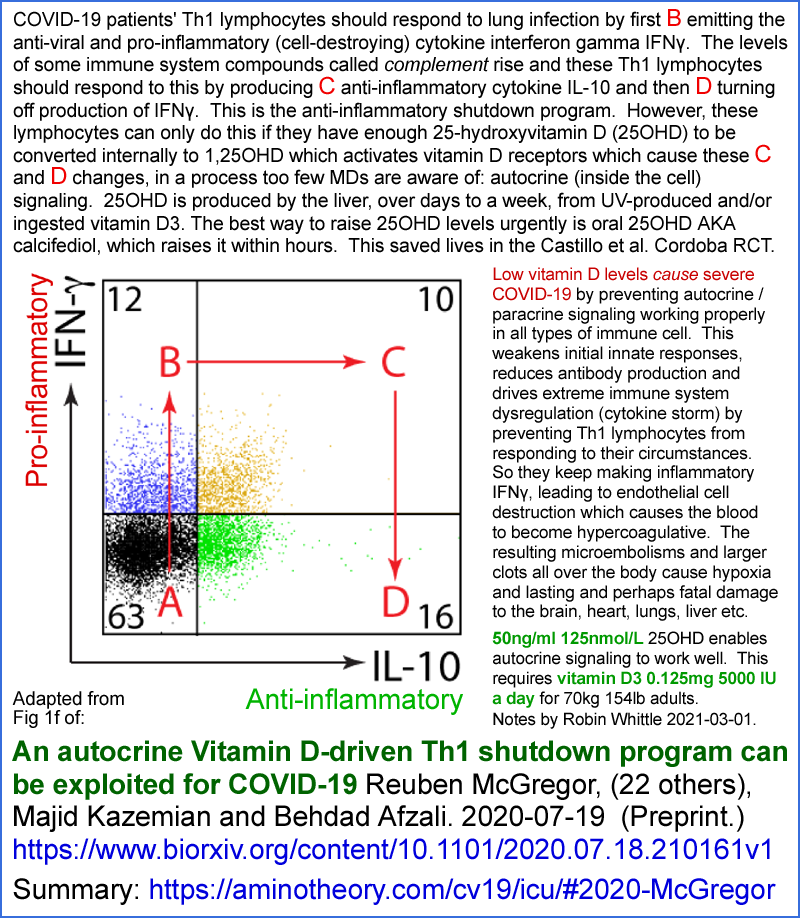
The wide positioning of the A, B, C and D on the above graph exaggerates the relative changes in the degree to which these two cytokines are created. (The horizontal and vertical dimensions of Fig 1f are for the rates at which the genes for these two cytokines are transcribed, which is approximately proportional to the rates at which the cytokines themselves are produced.)
I wanted to know the relative concentrations of the two cytokines produced by the cells between the B activated, inflammatory, state and the D shutdown, anti-inflammatory state. This can be estimated from each of two sets of data.
Firstly, the graphs in Fig 2C show the levels of the two cytokines produced when there is zero 1,25-dihydroxyvitamin D (hereafter 1,25(OH)2D) supplied to the cells, and then the levels when three different concentrations of 1,25(OH)2D are supplied. I judge the 10nM supply to be sufficient to fully activate the VDRs as they would be by normal, full, activation of the autocrine signaling system with sufficient 25-hydroxyvitamin D (hereafter 25(OH)D) supplied. So, for instance, for the top IFNγ curve of Fig 2c, I can divide the ~5900 pg/ml shutdown D level (4th red dot from the left) by the ~9,800 pg/ml B initial, inflammatory, level and get a D/B shutdown/inflammatory ratio of 0.6.
Secondly I can do the same for Fig S5d (in the separate Supplementary Materials PDF) which represents the same things, but this time in response to the experimenters supplying 25(OH)D so the real, already activated, intracrine signaling system can operate, producing its own 1,25(OH)2D to activate the cell's VDRs. Here, I chose the 3rd dot from the right, because I assumed, perhaps wrongly, that the 50nM (20ng/ml) 25(OH)D concentration was sufficient for the autocrine signaling system to operate fully. For IFNγ in this graph I estimate 7100 / 9100 (why not 9800 as before adding 1,25(OH)2D?) = 0.78.
I estimate that the D/B shutdown/inflammatory ratios are, from the two techniques respectively (first added 1,25(OH)2D and second added 25(OH)D) to be IFNγ 0.6 & 0.78, and IL-10 2.5 & 1.4 (ignoring the vertical scale discrepancy, which I assume is a typo). Neither of these experimental techniques tell us exactly what quantities of the two cytokines Th1 cells produce in-vivo, but it is the best we can do.
The normal, healthy, behaviour of Th1 lymphocytes to SARS-CoV-2 infection is as follows. (Those isolated from the lungs of severe COVID-19 sufferers did not transition to the anti-inflammatory program, and kept producing the pro-inflammatory IFNγ, presumably due to lack of sufficient 25(OH)D in those patient's bloodstream and therefore interstitial fluid, or whatever fluids the cells were bathed in.)
These Th1 cells have been activated (described below, perhaps before they multiplied in number) and so are in their pro-inflammatory B state (Fig 1f in the McGregor et al. preprint and in my infographic above), producing their higher level of IFNγ (WP) and their lower level of anti-inflammatory IL-10 (WP).
We are interested in understanding how they transition from this state, firstly to a temporary state C in Fig 1f, in which they produce high levels of both IFNγ and IL-10, and then to their final, anti-inflammatory, state D of low IFNγ production and high anti-inflammatory IL-10. This is the anti-inflammatory "shutdown program" referred to in the title of the preprint.
Messenger RNA analysis (scRNA-seq WP) of Th1 cells from the lungs of hospitalised COVID-19 patients and healthy controls revealed, in the patient's Th1 cells, elevated levels of mRNAs associated with the production of both IFNγ and complement.
Compared to the mRNA expression patterns in the T cells found in BALF (bronchoalveolar lavage fluid) of controls, patients' patterns were skewed to Th1 as opposed to the Th2 and Th7 lineages. However, no such skew was observed in T cells from peripheral blood:
High levels of complement production in Th1 cells has been observed in lung infections and specifically with SARS-CoV-2, which is known to particularly induce the production of complement factor C (C3). A fragment of this, C3b, binds to the CD46 receptors (WP) found on the plasma membranes of human (but not mouse) T cells. (2023-06-29: or is it just Th1 cells?) When all is working well, with sufficient 25(OH)D, the activation of the CD46 receptors causes the cell to shutdown from the initial pro-inflammatory B state to the final anti-inflammatory D state.
IL-10 mRNA in patients' BALF was found at about 1/4 the level it was found in controls, consistent with the hypothesis that Th1 cells in the lungs of hospitalised COVID-19 patients indeed did not initiate - or, at least, did not complete - their shutdown program.
Activation of CD46 receptors by C3b upregulated 24 transcription factors (TFs). (Genes for these 24 were found to have been transcribed to mRNAs at higher levels than without this CD46 activation.) One of these TFs was for the vitamin D receptor (VDR) gene and another was the CYP27B1 gene for the 1-hydroxylase enzyme, which converts intracellular 25(OH)D AKA calcifediol (diffused there from circulating 25(OH)D) into intracellular 1,25(OH)2D AKA calcitriol which binds to and activates the VDR.
So we see that this CD46 activation initiates the first steps of vitamin D intracrine signaling https://vitamindstopscovid.info/02-autocrine/ , while the other transcription factors drive other cellular responses in parallel to (if all works well) the effects of this intracrine signaling process, which might also involve this locally produced 1,25(OH)2D diffusing to nearby cells as a paracrine agent.
Although both vitamin D based intracrine and paracrine signaling had been previously described in Th1 cells, this research project set out to elucidate the molecular mechanisms and functional consequences of this, which were previously unknown. They did a great job!
Sidebar on vitamin D terminology and "vitamin D is a hormone":
In some articles, "vitamin D" means specifically vitamin D3 cholecalciferol, with 25(OH)D and 1,25(OH)2D being classed as "vitamin D metabolites". However, this makes no sense since the receptor universally known as the "vitamin D receptor", for all (or at least most) practical purposes, is only activated by 1,25(OH)2D. Some other articles use "vitamin D" to refer collectively to the three compounds: D3 cholecalciferol, 25(OH)D calcifediol and 1,25(OH)2D calcitriol. This makes sense to me, and this is how I use the term (2023-06-29: not any more!). Some articles use "vitamin D" to mean both these incompatible definitions, without any indication that this is invalid - which is very confusing.
This article classes the artificial compound 1,OHD alfacalcidiol [WP], which can be converted to 1,25(OH)2D by the 25-hydroxylase enzyme normally found in the liver (which normally converts D3 to 25(OH)D) as both an active metabolite of vitamin D, which I am pretty sure it is not, and simply as vitamin D, which it is not. Alphacalcidol is an analogue of "vitamin D", where this is the collective use of the term, since it is similar to D3 (an extra hydroxyl group at position 1) and can be converted to 1,25(OH)2D by adding another hydroxyl group at position 25. As far as I know, it is an artificial substance not found in-vivo.
Generally the article (and this is a preprint) uses the term "VitD" to refer to one of the three compounds, usually 1,25(OH)2D, but sometimes 25(OH)D, which I think is confusing. It would be better to refer to the specific compounds, in all cases.
1,25(OH)2D, when in circulation in the blood, acts as a hormone: endocrine cell-to-cell signaling between cells anywhere, or in many distant places in the body, with the compound being circulating in many parts of the body via the bloodstream.
This does not mean that 1,25(OH)2D is a hormone. It can act as a hormone when in circulation. Nor does it make any sense to state that "vitamin D" is a hormone, with this meaning either just D3 or collectively D3, 25(OH)D and 1,25(OH)2D.
However, this "vitamin D is a hormone" statement and this confused and confusing use of terminology is industry standard practice, since it is so common in vitamin D research articles, including a recent article co-written by the acknowledge leader of the field, Michael Holick:
Immunologic Effects of Vitamin D on Human Health and Disease Nipith Charoenngam, Michael F. Holick 2020-07-15 Nutrients 2020, 12(7), 2097 https://doi.org/10.3390/nu12072097 |
which I think is a great article, apart from these terminological problems.
(Page 5 to 6.) The researchers prepared activated* Th1 cells and treated them with 1,25(OH)2D to determine which genes were upregulated (296) and downregulated (157) by this robust VDR activation. (The precise details of these transcriptional - DNA to mRNA - changes would depend on the exact state of the cells, since there all sorts of subtleties in the direct mechanisms of transcription, and, for instance, acetylation of histones in ways which also affect the rate at which particular genes are transcribed.) Among these transcriptional changes were:
- Reduced transcription ("repressed") of the IFNG (interferon gamma) gene which, when
copied to mRNA, causes the protein making machinery (ribosomes) to
create IFNγ. So this change reduces the amount of IFNγ produced.
- Likewise, repression of the gene for the pro-inflammatory cytokine IL-17 [WP].
- Increased transcription ("induced") of the genes for IL-10, IL-6 (generally, but not always, a pro-inflammatory cytokine [WP]) and several transcription factor genes including JUN (for c-Jun [WP]), BACH2 (for the "broad complex-tramtrack-bric a brac and Cap'n'collar homology 2" protein [WP]) and STAT3 (Signal transducer and activator of transcription 3 [WP]). I discuss these further below. (It is 1AM and I can feel myself, very slowly, becoming a cell biologist - though I would not want to be examined on that BACH2 thang.)
The induction of IL-6 was a surprise to the researchers, since this is a frequently encountered and typically pro-inflammatory cytokine. However, as I best understand it, it seems that in these circumstances IL-6 is being produced for purposes of internal signaling (intracrine, I guess) rather than to stimulate high levels of inflammation in the vicinity of this cell.
The researchers repeated these tests and found the same patterns of gene transcription when, instead of directly adding 1,25(OH)2D, they added 25(OH)D (calcifediol). This shows that the activated cells, when they had enough 25(OH)D for the activation-created CYP27B1 1-hydroxylase enzyme molecules in their cytosol to convert to 1,25(OH)2D, which then binds to the VDR molecules there (also created by the activation). Then the bound complex of 1,25(OH)2D and VDR "migrates" (or does it simply diffuse?) to the nucleus, complexes with some other molecules (the retinoid X "receptor") and the resulting 3 molecule ensemble changes gene expression as expected so the cells transition successfully from their initial pro-inflammatory state B, through their pro- and anti-inflammatory state C, to their shutdown mode: anti-inflammatory state D.
We see from this that if all the cell's internal mechanisms are functioning normally, that successful transition from B to D would involve these external factors and these internal processes:
- High levels of complement
- perhaps created in part by the population of Th1 cells in the same
vicinity (millimetres, I guess) which are in the same initial B
state, (or is some or all of this complement, C3 protein at lest, made inside the Th1 cell??) lead to (by some
processes I am not trying to include in this summary) to the C3b part
of these complement compounds binding to the Th1 cell's CD46 receptors,
with their binding site on the outside of the cell's plasma
membrane.
- The activated CD46 receptor
alters (by means I am not trying to summarize) gene expression in many
ways, including by inducing (increasing the number of mRNA copies of)
the genes for the CYP27B1 1-hydroxylase enzyme and VDR.
These two of the many other gene expression altering changes caused by the activation of the CD46 receptors initiates this cell's vitamin D based autocrine (and potentially paracrine) signaling process. This process is initiated in a response to this particular cell's circumstances - it is not some kind of homeostasis-maintaining feedback loop. Each cell type which uses 25-hydroxyvitamin D based intracrine signaling has similar principles to steps 2 to 4, but the details of the initiating process and of which genes are induced and repressed are completely different between the cell types.
I think of this as the initiation step of this cell's vitamin D based intracrine signaling system. (I am leaving out various mRNA editing steps, if such steps occur with these genes - such as splicing removing introns - see post-transcriptional modification [WP].)
- These mRNAs leave the nucleus, get into the cytoplasm and there
direct ribosomes to make both the CYP27B1 1-hydroxylase enzyme and VDR
proteins. This is translation [WP],
but just of these two proteins (always the same for every cell which
uses 25-hydroxyvitamin D based intracrine / paracrine signaling).
(Meanwhile, other gene transcription changes caused directly by CD46
activation also result in more or less of other proteins being made,
but this is not part of the 25-hydroxyvitamin D based intracrine signaling process we
are focusing on at present.)
- Externally supplied 25-hydroxyvitamin D (AKA calcifediol and 25(OH)D) is necessary to the next step.
The cell cannot (generally <- added 2023-06-29) make its own
25(OH)D. Even if vitamin D3 is present in the interstitial fluid [WP]
(or perhaps the plasma, if the cell is in the bloodstream) it lacks the
25-hydroxylase enzyme to convert it to 25(OH)D. (2023-06-29 note,
this is not necessarily absolutely the case, since I recall that there
is evidence that some cells, other than those in the liver, can
hydroxylate vitamin D3 to 25(OH)D. For simplicity, here I assume
this is not the case to any significant degree for these Th1
lymphocytes.)
Generally we assume that UV-B produced or ingested D3 is converted, over days to a week or so, in the liver by this enzyme into 25(OH)D, where it goes into circulation in the blood plasma (more on binding proteins below).
As best I understand it, circulating 25(OH)D diffuses from the plasma into the interstitial fluid, without any particular active transporters or energy expenditure. From there, it diffuses - again without transmembrane transporters, energy expenditure or any other directional processes - from outside the cell's plasma membrane and into its cytosol. As far as I know, the 25(OH)D molecules are largely, but not entirely hydrophobic - only two hydroxyl groups and all the other outside parts of the molecule being oliophilic [WP] hydrogen atoms and small. I assume the molecule makes its own way across the plasma membrane. Can anyone provide more details or confirm this? (2023-06-29 note: I recall that kidney cells have an active transport arrangement to get 25(OH)D into the cells by carrying it attached to vitamin D binding protein, which is what the great majority of circulating 25(OH)D molecules are bound to.)
Inside the cell, 25(OH)D is subject to degradation by the 24-hydroxylase membrane, which is encoded by the CYP24A1 gene, the name of which is often applied to the enzyme itself. [WP]. (This is all basic vitamin D stuff - not specific to cells with vitamin D based intracrine / paracrine signaling systems.)
The 24-hydroxylase enzyme is inside the mitochondria [WP] and its molecular numbers, or at least its overall activity in the body, is upregulated by high 25(OH)D levels. (The diagram https://www.wikipathways.org/index.php/Pathway:WP1531
shows this enzyme degrading 25(OH)D, here referred to as calcidiol, and being upregulated only by parathyroid hormone and 1,25(OH)2D calcitriol, whether circulating or perhaps locally produced, rather than 25(OH)D. However, my potentially faulty understanding is that its activity is also driven by 25OHD itself, in a self-regulatory system which causes the curved D3 or 25(OH)D input to 25(OH)D level response in the Ekwaru et al. 2014 diagram: https://vitamindstopscovid.info/01-supp/a-ratios/ .)
This converts some 25(OH)D to 24,25(OH)D which is an irreversible operation, leading to the latter being metabolised and its components excreted. This is the primary, or perhaps sole, self-regulatory mechanism which tends to limit 25(OH)D levels (in the bloodstream and/or in individual cells) if there is a large input of vitamin D3 (and/or its UV-B creation) or (artificially) 25(OH)D into the body.
Secondly, this 24-hydroxylase enzyme also degrades 1,25(OH)2D, which has a much shorter half-life than 25(OH)D. (I guess this shorter half-life is due primarily to the greater affinity of 1,25(OH)2D for this 24-hydroxylase enzyme, but this is the limit of my knowledge and I don't have time now to dive into another rabbit hole to find out for sure.)
This externally supplied 25(OH)D (or that which remains subject to 24-hydroxylase activity AND it being already consumed as described next) finds its way to the active site of the 1 hydroxylase enzyme. The concentration of 25(OH)D in the cytosol is very low - so there is probably only one such molecule every 320 nanometres cubed (my rough calculations at https://vitamindstopscovid.info/02-intracrine/#03-minlev from which the next illustration is drawn) when the molecule, in red below, is only 0.2 nanometres long.
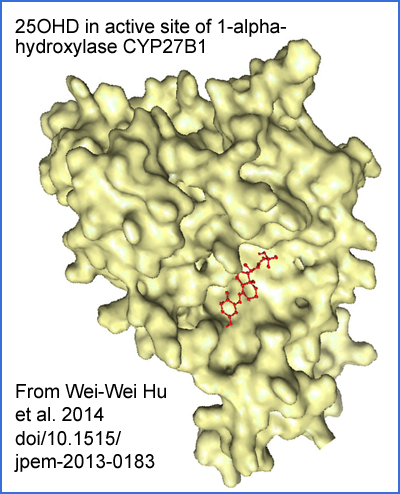
The 25(OH)D molecule also relies on thermal vibrations and some local electrostatic attraction to rotate it into the correct orientation and so slot into the active site correctly. I mention this to highlight that this reaction proceeds at a rate limited by the concentration of 25(OH)D, which is very low, and the likely still lower concentration of the 1-hydroxylase enzyme. (Cofactors are necessary for the 1-hydroxylation reaction, some of which are consumed in the process.)
The newly created 1,25(OH)2D molecule leaves the active site and the enzyme is ready for another 25(OH)D molecule to float into position.
The rate at which 1,25(OH)2D production proceeds is directly dependent on the 25(OH)D level in the cytosol, and this is reduced for every 25(OH)D molecule hydroxylated to 1,25(OH)2D, or lost to 24-hydroxylase.
For the intracrine signaling system to operate rapidly and fully, there needs to be a substantial rate of 25(OH)D to 1,25(OH)2D conversion, because the latter is short-lived. This means there needs to be a continual supply of 25(OH)D to the cell, by diffusion from the interstitial fluid (or perhaps directly from the plasma).
So the presence of adequate, externally supplied, 25(OH)D in the cytosol (which should be the case at all times - this presence not created by the CD46 initiation of this intracrine / paracrine signaling system) together with the newly created (by the initiation of the intracrine / paracrine signaling process) 1-hydroxylase enzyme leads directly to the outcome of this step: in-cytosol production of 1,25(OH)2D.
Some of this 1,25(OH)2D is degraded by 24-hydroxylase - over time, most of it which remains in the cell will presumably be degraded in this way. Some of this 1,25(OH)2D diffuses from the cell and may act on nearby cells as a paracrine agent.
Some of this 1,25(OH)2D drives the next step.
- The newly created 1,25(OH)2D molecules in the cytosol soon find themselves bound to the VDR, for which they have a very high affinity. There are a bunch of details regarding how the complex of the two "migrates"*
to the nucleus, binds with other things and (by various mechanisms I am
far from understanding - and I am not sure if anyone fully understands
them all) the presence of large numbers of bound VDR complexes alters
the transcription (AKA expression) of numerous genes. The exact
details of which genes are induced or suppressed, and to what degree,
depends entirely on the cell type.
* "Migration" is the usual term, but as far as I know there is no active, powered, directional mechanism for this. Can anyone improve my knowledge? I wrote "migrate" to be compatible with all I have read, but for now I assume the bound complex diffuses around the place randomly, with some of them getting into the nucleus.
This is the transcription phase of the intracrine signaling system.
There is some kind of degradation process for bound VDR complexes in the nucleus, so they don't accumulate and alter gene translation forever. I don't know the details, but I guess the VDR itself is retained and is free to diffuse back into the cytosol. However, there is surely a degradation process for the VDR as well, otherwise they would accumulate and this part of the intracrine signaling system would not be turned off when the initiating condition for the system is no longer active. The same goes for the 1-hydroxylase enzyme, which I assume is degraded in some way at a modest rate.
- The changed mRNA numbers for the various genes, by altering the protein produced by ribosomes, alters the function of the cell.
This is the final output of this entire vitamin D driven intracrine
signaling process for this particular type of cell.
mRNAs are rapidly degraded, and proteins (which may be exported or transformed in various ways) don't last forever. So for the autocrine signaling system to continue to function in its activated state, the original initiating stimulus (in the Th1 cell type, C3b binding to the CD46 receptors bound to, and sticking out of, the cell's plasma membrane) must continue to be present, 25(OH)D needs to be continually supplied, by diffusion, so 1,25(OH)2D production can continue. So there needs to be a continuing supply of 25(OH)D, by diffusion from the bloodstream into the cytosol of each cell.
The exact details by which IFNγ and IL-10 are produced by these Th1 lymphocytes is somewhat more complex than getting their mRNAs into the cytosol. More details of this were discovered by the researchers and are summarized below. However, these details do not alter the central role of 25-hydroxyvitamin D based intracrine signaling controlling the Th1 cells' production of these two crucially important cytokines other than that they also rely to some extent on the hydroxyvitamin D based intracrine signaling system also upregulating the transcription of the gene which produces IL-6.
This has been a rather detailed excursion into the molecular details of the central (common to all cell types which use it) mechanisms of 25-hydroxyvitamin D based intracrine signaling.
We need to think about this because at present (March 2021) the world is going to hell in a handbasket, in large part due to most humans having insufficient 25(OH)D in the cytoplasm of their immune cells to initially fight the SARS-CoV-2 infection, and in particular, in the context of humans' genetic predisposition to overly inflammatory immune responses due to lack of helminths:
If everyone had about 50ng/ml or more - twice is just fine (2023-06-29 note: for most people, there may be some people who have bad reactions to 100 ng/mL 250 nmol/L, but I have not yet been able to find all the best research) - 25(OH)D in their bloodstream, all these immune cell types, Th1 lymphocytes included, and all other cells in the body which use 25-hydroxyvitamin D based intracrine (and perhaps paracrine) signaling, would be working pretty well. Omega-3 and other nutrients such as magnesium, boron (#08-boron) and vitamin C are also important, but for now we focus on 25(OH)D, since this is the most important nutritional deficiency which clobbers all aspects of the immune system.
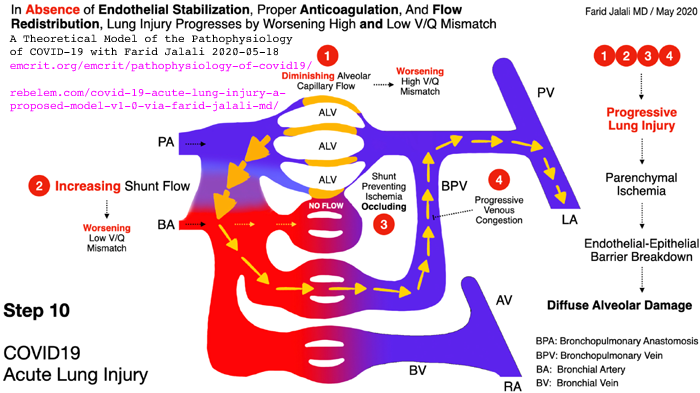
This McGregor et al. preprint concerns low 25(OH)D greatly increasing the (already genetically driven - see helminth material must mentioned) overly-inflammatory pattern of immune responses - and this response (perhaps just from these Th1 cells, but likely involving weak and dysregulated responses from all types of immune cell) is the primary driving reason why some COVID-19 sufferers progress to severe symptoms in their lungs, with high levels of endothelial damage, which causes the hypercoagulative blood which does the real damage with microembolisms and larger clots all through the body.
Inquiring minds now want to know how the (ideal, as far as we know ~50ng/ml 125nmol/L or more 25(OH)D blood levels relate to the levels of 25(OH)D in the cytoplasm of the Th1 cells (and all other cell types which also use vitamin D for autocrine / paracrine signaling). See my pages at the current site and at https://vitamindstopscovid.info for research which shows we need such levels, at least for intracrine signaling to work properly. (Some people, with auto-immune diseases, need two or three times these levels to significantly reduce or abolish their symptoms - here I am discussing most people, not these. See the McCullough et al. article mentioned above for examples.)
Looking at the experimenter's graphs for the IFNγ and IL-10 responses to 1,25(OH)2D (Fig 2C) and 25(OH)D (Fig S5d in the Supplementary data PDF), we see that most of the change occurs between concentrations of 0 and:
25(OH)D: (My guess, based on interpolating between the 0, 10 and 50nM levels.) 25nM with about half the action around 7nM.
nM == nano Mols == nmol/L = a billionth of a mole of molecules per litre.
1 nanogram per millilitre of D3, 25(OH)D or 1,25(OH)2D is the same concentration as 2.5nmol/L of these compounds. (This is ignoring the slightly heavier mass of the latter two.)
A Mole [WP] is 6.022 x 1023 molecules. 1 nanomol is 6 x 1014 molecules, so 1nM = 1nmol/L = 0.4ng/L means that for each molecule, there is 1.66 x 10-15 litres of water. This volume is a cube with sides 1.185 micrometres = 1185 nanometres, which is big for a molecule which is only 0.2 nanometres long. This is something like one 1mm long object in a home swimming pool.
The nM concentrations reported in these graphs are for the solution in which the researchers bathed the cells, and for now we assume reasonably free diffusion into the much smaller cell bodies - so these figures roughly represent the concentrations in the cytosol of these Th1 lymphocytes.
So it seems that to get the intracrine signaling system of these cells to respond fully, we need about 25nmol/L 25(OH)D in the cytosol, which is about 10ng/ml.
I think the discrepancy between this very approximate figure and the abovementioned ~50ng/mL for circulating 25(OH)D in the bloodstream can be accounted for roughly as follows:
- Most of the 25(OH)D in the blood plasma is bound tightly to vitamin D binding protein (DBP) molecules [WP],
which with 458 amino acids, dwarf the 25(OH)D molecule which is not much
bigger than a single amino acid. There is is considerable
individual and racial variation in the form of these DBP molecules and in
the concentration in which they are present in the plasma.
Although it is possible to measure the level of 25(OH)D which is not bound to DBP, this is not a commonly available from pathology labs, although such measurements might be more clinically relevant regarding the immune system.
DBP also binds D3 and the low level of circulating (hormonal) 1,25(OH)2D.
- Most of the 25(OH)D which is not bound to DBP molecules is bound - much less strongly, I recall - to albumin proteins [WP].
So the fraction of 25(OH)D available to diffuse into the interstitial fluid, and then into the cytosol of immune cells such as these Th1 lymphocytes, is the unbound fraction plus some part of the albumin-bound fraction.
- In any intracellular parts of the 25(OH)D molecules paths of diffusion from
the plasma to the cytosol of the Th1 lymphocytes (such as diffusing
through endothelial and other cells of capillaries), there will be some
losses due to the actions of the 24-hydroxylase enzyme.
So lets think about D3 intakes and these low 25(OH)D levels for a moment. For 70kg adults, 0.125mg 5000IU D3 a day maintains about 50ng/ml. This is 1 gram every 22 years. These patients lower 25(OH)D levels of 4 to 20ng/ml probably result from much lower total quantities of D3 from UV-B, food and few, if any, supplements. Let's say their D3 input was 0.01mg 400IU a day, with some patients, in recent years at least, with even less, due to being aged, being indoors, having dark skin, covering their bodies almost entirely when outside etc. The UK government recommends 0.01mg 400IU D3 a day for adults - in winter only.
These people would generally not be patients in hospital with COVID-19 if they had been supplementing 0.125mg D3 a day (or more according to bodyweight, with more for people suffering from obesity: https://vitamindstopscovid.info/01-supp/ ). 2023-06:29: see the chart of vitamin D3 supplemental intake quantities, as ratios of body weight, at: https://brownstone.org/articles/vitamin-d-everything-you-need-to-know/ .
So this severe ill-health, and numerous other aspects of ill health, has been in large part caused by these patients having disastrously low D3 intakes (though generally about enough to ensure they don't get rickets or unusual levels of osteoporosis), of about 1 gram every 100,000 days = 274 years. Pharma grade D3 costs about USD$2.50 a gram ex factory in 1kg quantities. (That said, UK intracrine signaling pioneer Martin Hewison states in a 2021-03-03 video that the UK is the vitamin D deficiency centre of the world, and that rickets is not uncommon in certain communities - presumably of people with dark skin and/or who avoid direct sun exposure of their skin.)
This global epidemic of vitamin D deficiency has been running for decades and centuries. (2023-06-23 update: since humans started living in Europe and especially northern Europe, eventually migrating to various other locations far from the equator.) Without it, there would be little or no need for the disastrous lockdowns, social distancing, economic and travel shutdowns etc. which governments have imposed to combat COVID-19, and surely will continue to impose in the years as vaccine manufacturers (and populations of entire countries) play cat-and-mouse, or whack-a-mole, with increasingly efficient viral variants which are also under strong selection pressure to evolve avoidance of existing infection- and vaccine-induced immune responses.
Back to the research article!
This concludes the section of my summary and discussion which is most relevant to 25-hydroxyvitamin D based intracrine signaling in Th1 lymphocytes. What follows is an attempt to summarize some of the other molecular details of exactly how IFNγ, IL-10 and IL-6 production is regulated in these cells, within the context of these Th1 cells' 25-hydroxyvitamin D based intracrine / paracrine signaling system.
Again, please remember this is the best effort of an electronic technician to understand and describe complex cell-biology experimentation.
The researchers noticed that IL-10 production was proportional to IL-6 production. They added IL-6 to cells which had their CD46 receptors stimulated, and so the 25-hydroxyvitamin D based intracrine signaling system had been initiated, but with the cells remained stuck in their pro-inflammatory B state due to there being insufficient 25(OH)D in the cytosol. (With sufficient 25(OH)D they would have converted some of it to 1,25(OH)2D, completed the autocrine signaling pathway and transitioned through C to the anti-inflammatory D state.) This caused the cells to produce pro-inflammatory IL-17, which had been reported by other researchers. This seems clinically relevant to me, since this is an additional mechanism for pro-inflammatory dysregulation in patients with low 25(OH)D levels, IL-6 being at high levels in all these COVID-19 lung infections.
Both c-JUN and STAT3 transcription factors were produced by the full operation of the 25-hydroxyvitamin D based intracrine signaling system (leading to state D). c-JUN phosphorylation [WP] was also driven directly by this. STAT3 phosphorylation was dependent not on this completion of the intracrine signaling pathway, but on the presence of IL-6, the production of which was directly caused by the completion of the intracrine signaling system. The researchers confirmed that the IL-6 receptor was necessary for this process by introducing an IL-6 receptor blocker Tocilizumab. (Imagine the dreams of the people whose job it is to come up with these names for pharmaceuticals.)
Since this IL-6 was, at least in part, produced inside the cell, and activates receptors in the same cell, this is a separate IL-6 based intracrine pathway with these IL-6 molecules acting as an intracrine agent. To the extent that some of the IL-6 which activates these receptors was produced in nearby cells (Th1 or other types), those IL-6 molecules are acting as paracrine agents.
With further evidence from the cells of two patients with a rare STAT3 mutation (!) the researchers concluded:
VitD [they mean completion of the 25-hydroxyvitamin D based intracrine signaling response to high levels of complement, which is only possible with sufficient 25(OH)D] induces STAT3 and IL-6, and IL-6 phosphorylates STAT3 to promote production of [anti-inflammatory] IL-10. |
Page 7 to 8 describes the researchers' investigation of the molecular details of how VDR activation (which will transition these cells from the B state, through C to D) works, such as by the VDR, complexed to other molecules, affecting the degree to which histones [WP] are acetylated [WP] in particular places. The 46 chromosomes add up to 1.8 metres of DNA which is wound around much smaller histone molecules. This typically tight winding impedes access to most of the DNA by the enzymes which can copy its information into messenger RNA molecules (mRNAs). Acetylation in particular parts of the histone enables looser DNA winding and therefore greater access of these enzymes to particular parts of the DNA - including the genes which, in this particular cell type, VDR activation increases the transcription of. I don't clearly understand their observations regarding c-Jun, BACH2 and STAT3 transcription factors, their phosphorylation and super enhancer structures arising from histone acetylation.
BACH2 - a transcription factor whose production is increased "by vitamin D" (meaning the successful completion of the 25-hydroxyvitamin D based intracrine process leading to state D) - is crucial to the increased transcription of other genes by this process, including the gene for the IL-6 receptor which, as as noted previously, is an essential link in the chain of IL-6 based intracrine / paracrine signaling which leads to the production of anti-inflammatory IL-10.
| Expression
of VitD-repressed genes . . . was significantly higher in
bronchoalveolar fluid Th cells of patients with COVID-19 than
healthy controls. |
So the Th1 cells from hospitalised COVID-19 patients did not have their (high levels of complement driven) CD46 activated 25-hydroxyvitamin D based intracrine signaling function run to completion (from state B through C to D), presumably due to lack of 25(OH)D, leading to little or no production of intracrine agent 1,25(OH)2D, and so little or no activation of VDRs which, if activated, would have repressed the transcription of these genes.
Here endeth the lesson. There will be a test on Friday!